 Print This Post
Print This PostThe FDA’s controversial approval of aducanumab hinged on the premise that clearance of amyloid would be “reasonably likely” to bestow a cognitive benefit. Data presented at the Alzheimer’s Association International Conference (AAIC), held July 26-30 in Denver, Colorado, and online, support the idea that two other antibodies could clear that low bar, as well. Scientists from Eli Lilly reported that the plaque-dissolving strength of donanemab, an antibody trained against forms of Aβ detectable only in plaques, tracked closely with plummeting plasma p-tau217. Weaving their data into a disease-progression model that had been generated from past trial data, they claimed that the amyloid- and tau-lowering effects of the drug correlated with a slowing of cognitive decline. Separately, data from lecanemab’s Phase 2 trial and open-label extension studies provided yet more support for that antibody’s disease-modifying effect, despite the travails that have beset its path through clinical development.
- In a Phase 2 trial of donanemab, Aβ plaques and plasma p-tau217 fell in unison.
- These shifts correlated with slower cognitive decline decline based on a disease progression model generated with historical controls.
- After amyloid clearance and switch to placebo, plaque levels remained low for at least a year.
“I was mightily encouraged by the data presented by Lilly and Eisai, which gave further support for the FDA assertion that there was a ‘reasonable likelihood’ that lowering the Aβ-load would result in clinical benefit,” commented Colin Masters of the University of Melbourne, Australia.
Both donanemab and lecanemab have received breakthrough therapy status from the FDA. Similarly to aducanumab, this means that their sponsors could apply for accelerated approval based primarily on changes in surrogate biomarkers that demonstrate amyloid reduction. Both Lilly and Biogen/Eisai have announced plans to use their Phase 2 data to apply for accelerated approval of donanemab and lecanemab, respectively. Lilly will file later this year (see Endpoints News).
“Lilly is in the catbird seat with donanemab,” said Lon Schneider of the University of Southern California in Los Angeles. He noted that Lilly’s Phase 2 trial met its primary endpoint, and its biomarker data showing both amyloid and tau reduction were more convincing than Biogen’s on aducanumab. Even so, Schneider continues to disagree with the new precedent set by the FDA, and favors that Phase 3 clinical efficacy be demonstrated prior to approval of any therapeutic antibody.
Alzforum covered results of the Phase 2 donanemab trial—called TRAILBLAZER—in March, just as the data was also published (Mar 2021 conference news; Mintun et al., 2021). At AAIC, John Sims and Mark Mintun of Lilly reported fresh analyses since then of the amyloid and tau biomarker data, respectively.
The trial enrolled 257 participants who had early symptomatic AD, amyloid in their brains, and—notably—an intermediate level of neurofibrillary tangles based on PET scan. After an initial period, when 131 volunteers randomized to the treatment group gradually received higher and higher doses of donanemab, the trial settled in on monthly infusions of 1,400 mg donanemab. This was given until a person’s amyloid burden dropped below 25 centiloids—the level in healthy young controls—at which point the dose was lowered to 700 mg. If amyloid fell below 11 centiloids, or below 25 for two consecutive scans, the person was switched to placebo.
As previously reported, the 76-week trial met its primary cognitive endpoint, showing a 32 percent slowing of decline on the Integrated Alzheimer’s Disease Rating Scale (iADRS). By 24 weeks, donanemab had completely cleared plaques in 40 percent of participants in the treatment group; by the trial’s end, 68 percent had reached normal levels.
At AAIC, Sims broke down that amyloid reduction data further. Most participants in the treatment group had a rapid reduction in amyloid over the first 24 weeks. As might be expected, the amount of amyloid they shed over those first six months depended upon how much they started with, Sims reported. In other words, people with higher levels of amyloid at baseline lost more than those who started with less. Still, they took longer to dip into the realm of healthy young controls.
Sims reported that once a person’s amyloid complete cleared, their levels stayed down for the remainder of the trial. Among the participants with “deep amyloid clearance,” i.e., amyloid levels below 11 centiloids, and who were switched to placebo by 24 weeks, amyloid burden crept up only slowly by 76 weeks, barely cresting 11 centiloids, on average. At this rate, it would take 14 years for amyloid to accumulate back to baseline level for this group, or about 90 centiloids, Sims reported (see image below).
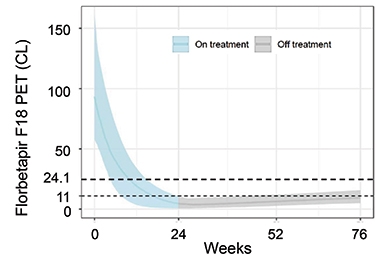
Get Out, Stay Out. Among people who had complete removal of amyloid plaques by 24 weeks, and were switched to placebo at that time, amyloid burden stayed within the range of healthy young controls (less than 25 centiloids) for the remainder of the trial. [Courtesy of Eli Lilly.]
How did this rapid amyloid clearance influence downstream parts of the AD cascade, i.e., tau tangles and cognitive decline? All participants underwent flortaucipir-PET scans at baseline and 76 weeks. Sims reported that compared to the placebo group, those who had had complete clearance of amyloid by 24 weeks had a significant dip in tangle burden in the temporal, parietal, and frontal lobes by the end of the trial. People who had had partial amyloid clearance by 24 weeks also had significant drops in tau tracer uptake, at least in the parietal and frontal lobes. “These findings link us back to the concept of AD as an amyloid-induced tauopathy,” Sims said.
Would riddance of amyloid also track with slowing of cognitive decline? Among all participants, including those in the placebo group, the percent change in amyloid burden by 24 weeks had no significant correlation with the change in iADRS scores between baseline and 52, 64, and 76 weeks, though there was a trend toward slower cognitive decline among those who had cleared the most amyloid.
Given the small numbers of participants in the Phase 2 trial, Sims further analyzed the trial data using a disease-progression model developed at Pfizer that was based on data from nearly 5,000 controls (Conrado et al., 2014). The Coalition Against Major Diseases had gathered this control data, including from the placebo arms of 15 AD clinical trials conducted between the 1990s and 2010 on people who had mild to moderate AD (Jul 2013 news; Dec 2010 news). The model calculates trajectories of disease progression—as gauged by ADAS-Cog scores—among populations with different characteristics, such as ApoE4 carriers or noncarriers.
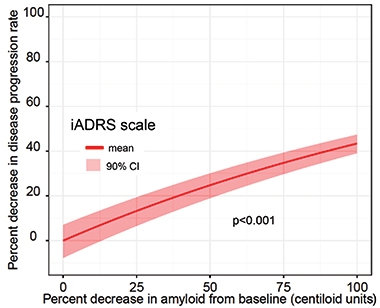
Model Comparison. By plugging percent amyloid reduction into the Conrado model, Sims found a statistically significant relationship between amyloid clearance and slowing of disease progression. [Courtesy of Eli Lilly.]
When Sims plugged data from TRAILBLAZER into the model, it estimated that donanemab slowed cognitive decline by 28 percent among all participants in the treatment group, and by 42 percent among APOE4 carriers. The findings jibe with the 32 percent slowing of cognitive decline that the researchers had previously reported, though the model lowered the p value, at less than 0.001. The model also rendered a statistically significant link between amyloid reduction and disease slowing.
Schneider pointed out that the model used data from people who had mild to moderate AD, whereas TRAILBLAZER selected participants with MCI to mild AD. “So there’s a good chance that many of the inputs from the TRAILBLAZER trial are where the model is least accurate,” Schneider said. Still, he and other researchers felt that the Lilly researchers were doing their best with the limited data available.
Plasma p-tau217 Tracks with Amyloid Drop
Mintun’s presentation focused squarely on freshly garnered data from Lilly’s in-house plasma p-tau217 assay. Lilly originally developed the assay on the Meso Scale Discovery platform, which works like an ELISA. At AAIC, Mintun debuted the assay’s adaptation to the more sensitive Simoa platform. Several groups have reported that p-tau217 in the CSF and plasma rise early in the disease trajectory and correlate with amyloid and tangle load (Apr 2020 conference news; Jul 2020 conference news). In a first for the field, Mintun reported how plasma p-tau217 levels changed in response to treatment.
At baseline, plasma p-tau217 correlated with baseline amyloid-PET and tau-PET measures as reported previously. This may seem like a no-brainer, but the result is striking considering that trial enrollment was restricted to people with an intermediate level of tau tracer uptake, thus limiting the dynamic range of PET measurements at baseline, Mintun said.
What happened to p-tau217 as brain amyloid plummeted in response to donanemab treatment? In short, it followed suit. Mintun reported that by 12 weeks, plasma p-tau217 was already significantly lower in the donanemab group compared to placebo. Levels continued to fall at every time point, while statistical significance strengthened. By the end of the trial, plasma p-tau217 had fallen by 24 percent in the treatment group, and risen by 6 percent in the placebo group. The latter rise is on par with that expected during disease progression, Mintun said.

Plasma p-Tau217 Plummets. In response to donanemab treatment, amyloid plaque burden (left) and plasma p-tau217 dropped throughout the trial. [Courtesy of Eli Lilly.]
Notably, at 24, 52, and 76 weeks, the drop in plasma p-tau217 from baseline was strongly tied to reduction in amyloid. In addition, reduction in p-tau217 was linked to reduction in tau tangles, according to tau-PET, between the baseline and the end of the trial.
Mintun also applied the p-tau217 data to the disease progression model that Sims had employed. This cast change in plasma p-tau217 as a significant predictor of slowed disease progression.
Henrik Zetterberg of the University of Gothenburg in Sweden called the responsiveness of plasma p-tau217 to donanemab treatment “a Hallelujah moment,” noting that this was the first time rigorous plasma p-tau biomarker data had been presented in the context of a clinical trial. He said the findings provide strong support for the amyloid cascade hypothesis. That p-tau217 dropped so quickly in response to amyloid removal supports the idea that neurons actively release phospho-tau in response to exposure to amyloid. “If we reduce amyloid exposure, we would expect to see a very quick decrease in phospho-tau secretion,” Zetterberg said. “And that’s exactly what we see.”
This data was also a meeting highlight for Oskar Hansson of Lund University, Sweden. “These important results clearly indicate that p-tau217 in plasma might be used as an easily accessible and cost-effective marker revealing effects of novel treatments on the levels of amyloid-β fibrils in the brain,” Hansson wrote. However, he emphasized that it is not yet known whether plasma p-tau217 can be used as a surrogate biomarker of clinical efficacy. “We simply do not yet know if the reduction of extracellular levels of soluble p-tau is an epiphenomenon merely associated with a reduction in β-amyloid fibrils in the brain, or a key event that will always lead to diminished tau aggregation, less neuronal dysfunction, and reduced degeneration, independently of the mechanism causing the reduced levels of extracellular p-tau to begin with.”
Paul Aisen of the University of Southern California in San Diego made a similar point. “The treatment-related fall in p-tau217 must still be linked to clinical benefit as validation of its utility in guiding therapy,” he wrote. “But if it can be validated, this assay will be an enormously useful tool in the drug-development process.”
David Morgan of Michigan State University in Grand Rapids, who attended the meeting in person, asked Mintun if and when they would measure other blood biomarkers that could hint at a neuroprotective effect of treatment, such as neurofilament light (NfL) and glial fibrillary acidic protein (GFAP). Absent solid cognitive data, perhaps these markers could get closer to addressing the question of efficacy, Morgan told Alzforum. Mintun said that they intend to measure other markers, but optimizing the p-tau217 assay on the sensitive Simoa platform had been top priority leading up to the meeting.
Zetterberg believes this effort may have been well worth it. Given the vanishingly small concentration of p-tau217 in the plasma, the added boost of sensitivity may have been crucial for detecting the treatment effect. Zetterberg said that while he looks forward to seeing how neurodegenerative biomarkers respond to donanemab treatment, the data could prove difficult to interpret. “As microglia are recruited to donanemab-bound plaques, GFAP levels might go wild,” he said. NfL could even rise for a time, he added, as glial cells nosh on detritus from amyloid-exposed neurons. Zetterberg acknowledged that while potentially interesting and important, complex biomarker data might be the last thing Lilly wants while seeking accelerated approval of donanemab by the FDA.
The most exciting implication of the donanemab data, Zetterberg said, was the possibility of treating patients for only a few months to remove plaques. Then, plasma p-tau markers could be used to monitor patients for the return of plaques, at which point they could take another round of treatment, he suggested. Zetterberg was heartened by the data showing that neither Aβ plaques nor plasma p-tau217 appeared to rise within a year after donanemab treatment stopped.
If approved, “amyloid plaque removal” could become a common treatment for people in their golden years who go to their doctors with cognitive complaints, Schneider said.
What about for cognitively normal people who have amyloid plaques? Could donanemab be approved for them? For now, aducanumab is only conditionally approved for people in the early symptomatic stages of AD. Both donanemab and lecanemab trials have included only this population as well. That will soon change, however, when TRAILBLAZER-ALZ3 begins. This Phase 3 trial will test donanemab in cognitively unimpaired people at risk for AD, and will use plasma p-tau217 to select participants (Jul 2021 news).
Lecanemab is also being put to the test in people with preclinical AD in the AHEAD3-45 trial, which is enrolling participants. At AAIC, Chad Swanson of Eisai presented data from the Phase 2 study of lecanemab and an 18-month open label extension.—Jessica Shugart
Shared from the Alz Forum – https://www.alzforum.org/news/conference-coverage/donanemab-plaques-plummet-donanemab-they-stay-away#comment-41786





